




# 前言 #
欧盟体外诊断医疗器械新法规(IVDR) EU 2017/746 于2017年5月颁布,标志着98/79/EEC 废止的五年过渡期开始。
2022年5月26日是IVDR的实施日期,标志着法规交替过渡期的结束。
本文从IVDD指令到IVDR的最主要变化,到发证机构的现状,欧盟政府的调整,以及厂家的应对几方面进行梳理,希望对业内的朋友有所启发和帮助。
98/79/EEC IVDD 的“天生缺陷”
众所周知,98/79/EEC的分类规则仅有寥寥数行,如下图:

指令原文里面只有List A和 List B两种分类,被囊括的产品更是极少,相比数量庞大的IVDD品类来说,这样的分类只导致一种结果:
90%的IVD产品没被纳入进需要NB介入机构发证的范畴,也就是我们俗称的others类产品。
EU 2017/746 IVDR的“大刀阔斧”
IVDR在产品分类方面参照了2017/745 MDR 的分类规则,将IVD产品分成了Class A,B,C,D 4类,把具体的一类产品归在一个分类规则下,比如:
检测是否存在或显露传染性因子, 其会导致危及生命的疾病, 并且具有高的或可疑的传播风险。
这类产品,就属于IVDR 风险最 高的Class D类产品。
IVDR的分类规则不再像IVDD指令那样的划分具体的产品个体,比如乙肝产品就划分为List A类产品。
NB机构的“力不从心”
在近几年的时间里,大量的IVD厂家的产品需要寻求发证机构的CE审核及发证服务,但是发证机构本身IVD专家资源的匮乏,IVDR发证机构的稀缺(到目前为止尚只有7家已经获取IVDR资质的NB机构),加之整个IVD行业由于疫情的井喷式增长以及新法规的实施的叠加效应,导致了NB机构的“力不从心”。
欧盟政府的“被逼无奈”
作为欧洲NB机构的监管方,欧盟政府就NB机构运力不足的问题,在2022年1月25日,由欧洲议会和欧盟委员会发布REGULATION (EU)2022/112,确定了IVDR 过渡期延期事宜。
依据发布的REGULATION (EU) 2022/112,具体延期如下:
-过渡期总体由 2024.5.27 推迟到 2025.5.27;
-在2022.5.26之前完成了NB发证的IVDD下List A, List B和Self-test产品,可继续销售至2025.5.26;
-对于 IVDD 下 other 类产品,在 2022.5.26 之前完成了IVDD符合性声明(欧盟注册)的,在如下日期之前仍可上市销售:
・ Class D(IVDR下)2025.5.26;
・ Class C(IVDR下)2026.5.26;
・ Class B(IVDR下)2027.5.26;
・ Class A无菌(IVDR下)2027.5.26。
-对于 IVDD 下 other 类产品,在 2022.5.26 之后完成IVDD符合性声明(欧盟注册)的,在如下日期之前可上市销售:
・ Class D(IVDR下)2026.5.26;
・ Class C(IVDR下)2027.5.26;
・ Class B(IVDR下)2028.5.26;
・ Class A无菌(IVDR下)2028.5.26。
IVDR 新政并没有推迟,还是 2022.5.26 实施;PMS、警戒系统、经济运营商注册这些要求,依旧自2022.5.26立即实施;由此,企业需及时更新QMS,同时依据IVDR要求实施企业注册、产品注册等。
IVD厂家的“未雨绸缪”
很多IVD厂家前期在联系NB发证机构之时可能都会被告知机构不受理,但是发证机构受理与否永远是一个动态的不可预知的过程,IVDR产品众多,相应的性能检测,可用性检测,临床实验都会花费较长的时间,IVD厂家比较恰当的做法就是在等待机构的同时把相应的前置条件先完成,这样,一旦发证机构受理,就可以赶上头班车,大大节约等待和准备的时间。
已获IVDR授权的公告机构,共计7家 (信息来源:NANDO)

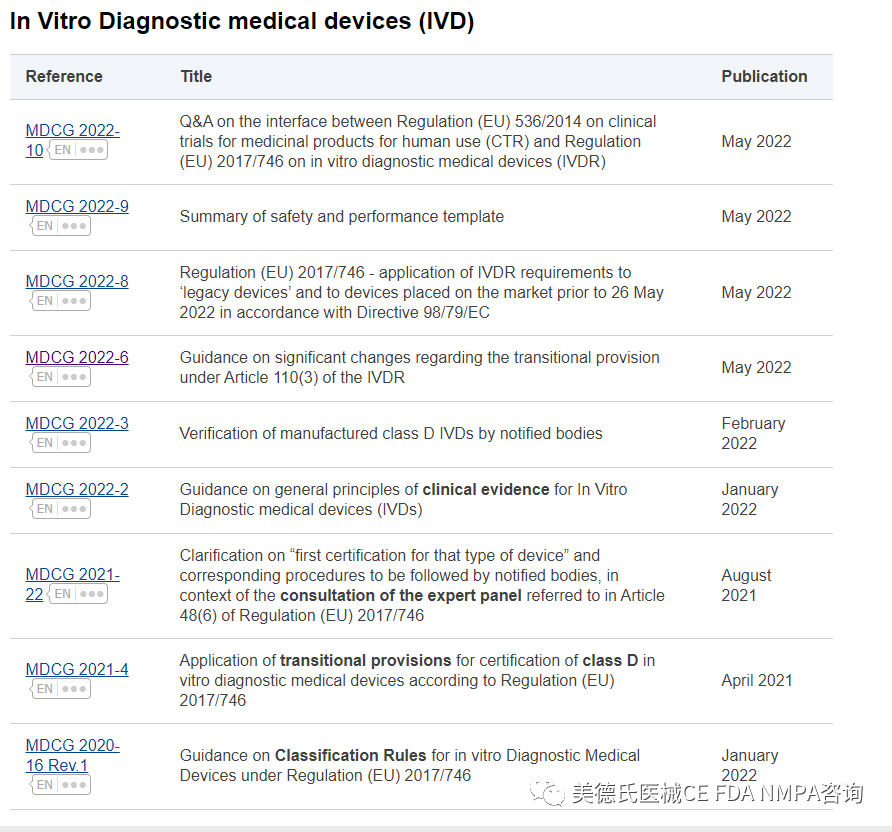
已发布的MDCG IVDR指南文件
本文相关链接
延期法案:
https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32022R0112&from=EN
MDCG指南下载链接:
https://ec.europa.eu/health/md_sector/new_regulations/guidance_en
NANDO公告机构数据库:
https://ec.europa.eu/growth/tools-databases/nando/
美德氏医械服务范围
质量管理体系服务
服务 | 内容 |
| ISO13485 | ISO 13485的质量体系的培训、建立及运行辅导 |
| FDA | FDA CFR820 的质量体系的培训、建立及运行辅导 |
| MDSAP | MDSAP的质量体系的培训、建立及运行辅导 |
| NMPA | NMPA(GMP、GSP)的质量体系的培训、建立及运行辅导 |
| 质量体系日常维护服务 | FDA820的不符合项以及警告信、欧盟的CAPA、流程改进、质量体系维护的外包、供应商审核 |
法规合规及产品注册服务
| 服务 | 内容 |
| 欧盟市场准入整体解决方案 | 包括CE技术文档撰写、辅导、测试、认证全套方案。还包括欧代服务、欧盟FSC、ISO14971 风险分析、临床评价、灭菌、软件周期、可用性等欧盟合规的咨询与服务 |
| 美国市场准入整体解决方案 | 包括510K文档撰写与认证,产品列名、工厂注册、美国代理人、UDI的合规咨询与服务 |
| 中国市场准入整体解决方案 | 包括NMPA文档撰写与注册,生产许可证、中国FSC、医疗器械广告审核的合规咨询与服务 |
| 其他国家的认证注册咨询服务 | 包括全球法规注册咨询服务,如澳大利亚、新西兰、加拿大、巴西、俄罗斯、日本、韩国等全球国家的注册咨询服务 |
医疗器械法规培训
服务 | 内容 |
| 国内外法规培训精讲 | MDR 2017/745/EU 法规培训 |
| IVDR 2017/746/EU 法规培训 | |
| MDR临床实验方案设计培训 | |
| IVDR临床实验,性能实验方案设计培训 | |
| ISO14971-2019 医疗器械风险管理培训 | |
| 《ISO13485-2016医疗器械 质量管理体系用于法规的要求》深度解读 | |
| 产品设计开发,产品可用性工程培训 | |
| ISO 11135 11137 灭菌培训 | |
| 其他定制式企业培训 |