





法规框架
在美国销售的医疗器械应遵守《联邦食品药品和化妆品法案》(FD&C Act)的法规控制以及《标题21-联邦法规法典》(21 CFR)第1-58、800-1299部分的法规 。

美国医疗器械的法规历史
1976: Medical Device Amendments to the FD&C Act
-旨在确保医疗器械的安全性和有效性;
-建立了一个基于产品风险的三级分类系统;
-建立了新医疗器械的上市法规路径:上市前审批PMA和上市前通知510(k);
-建立了新的研究类医疗器械(IDE)的法规路径;
-建立了后市场的要求,包括场地注册,产品列名,不良事件的汇报,GMP等;
1990: Safe Medical Devices Act (SMDA)
-加强后市场的监管;
-定义了510(k) 中实质性等同的含义。
1997: Food and Drug Administration Modernization Act (FDAMA)
-创建了第三方认证的选项,可以对某些设备进行上市前的初步审查
-建立了De Novo计划,通过该计划,新型中低风险设备可以分类为I级或II级,而不是自动将其分类为III级
2002: Medical Device User Fee and Modernization Act (MDUFMA)
-授予FDA收取针对某些医疗器械上市前提交的使用费,以帮助FDA提高医疗器械提交审查的效率,质量和可预测性
-实施了小型企业确定(SBD)计划,以降低合格的小型企业的上市前批准费用
-为某些上市前提交的决定制定FDA绩效目标
-建立了针对“再加工”设备的新法规要求
-医疗器械公司的授权电子注册
2007: Food and Drug Administration Amendments Act (FDAAA)
-重新修订了医疗器械使用费,包括缩短了上市前审查时间;
-要求所有注册和列名均以电子方式执行;
-要求FDA为医疗设备建立的设备识别(UDI)系统,以要求设备标签带有的标识符。
2016: 21st Century Cures Act
-通过定义不能作为设备进行监管的医疗软件类别,阐明了如何监管某些数字保健产品。
2017: Food and Drug Administration Reauthorization Act (FDARA)
-重新修订了医疗器械使用费计划;
-授权了针对设备企业的基于风险的检查计划,并规定了与设备企业审核相关的其他流程改进。
FDA对于医疗器械的定义
设备,仪器,工具,机器,配件,植入物,体外试剂或其他类似或相关的物品,包括以下组成部分或附件:
在官方国家处方或美国药典或其任何补充中认可的药物,
- 旨在用于人类或其他动物的疾病或其他状况的诊断或治愈,缓解,治疗或预防疾病的用途,或
- 旨在影响人类或其他动物的身体的结构或任何功能,并且无法通过人类或其他动物的身体内部或之上的化学作用实现其主要预期目的,并且
它不能通过在人或其他动物体内或对人体的化学作用来达到其主要预期目的,并且不依赖于通过代谢来实现其主要预期目的。
美国医疗器械的主管机构
美国食品药品监督管理局(FDA)对食品和药品的监督始于1906年,罗斯福总统签署了《联邦食品和药品法》。从那时起,国会扩大了FDA在保护和促进人类和兽药,生物产品,医疗设备和辐射发射产品,人类和动物食品以及化妆品中的作用。医疗器械的主管部门为美国食品和药品管理局(FDA)下属的器械和放射健康中心(CDRH)。

医疗器械的上市路径

-确定器械分类
美国食品药品监督管理局(FDA)已为大约1,700种不同通用类型的设备建立了分类,设备分类是基于风险的,也就是说,设备对患者和/或用户造成的风险是分配给设备的类别中的主要因素。I类为风险低的设备,III类为风险大的设备。
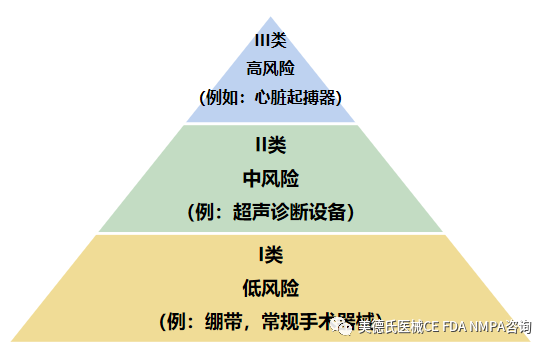
-如何寻找器械分类?
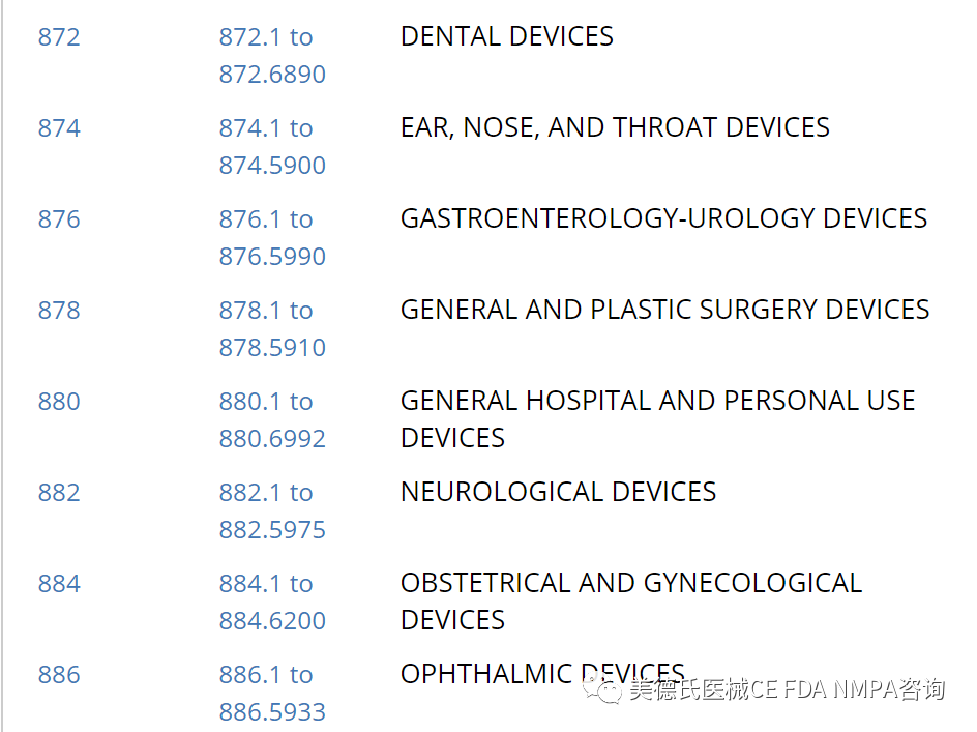
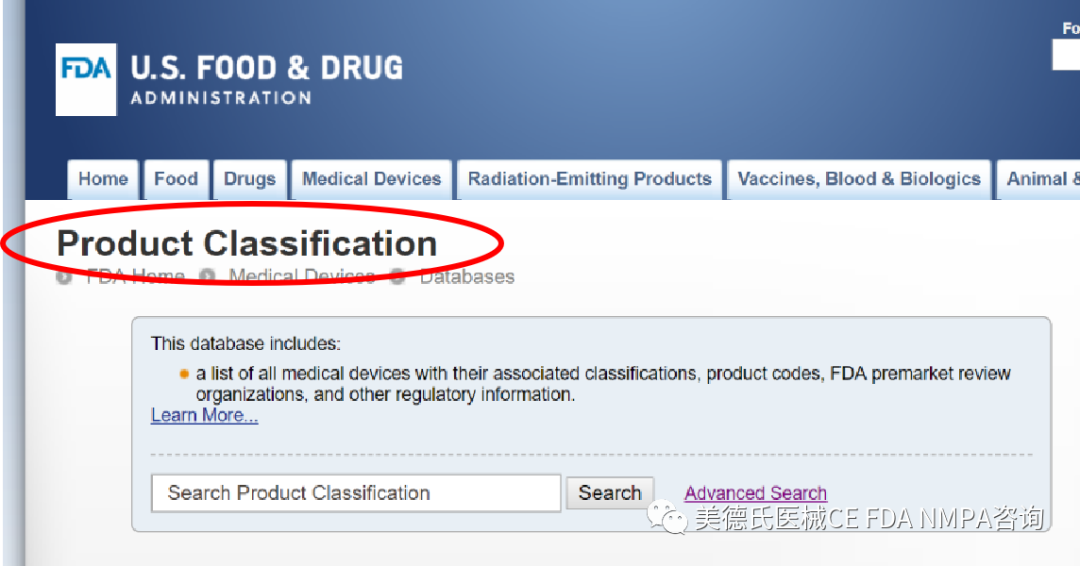
-选择合适的上市前路径
在对设备进行分类之后,选择该法规所需的上市前路径提交文件。上市前常见的提交方式包括:
510(k)(上市前通知)
PMA(上市前批准)
De Novo(自动III级的评估)
HDE(人道主义设备豁免)
-510(k)(上市前通知)
510(k)是在医疗器械上市前递交给FDA的一套文件,来证明所申请的产品与已在美国上市的产品是实质等同(SE)的。申请人在拿到FDA的批准信之前,该医疗器械是不能在美国上市的。
某些I类设备不受上市前通知和/或良好生产规范的约束。大约572或74%的I类设备免于上市前通知流程。
大部分II类产品和部分I类产品是需要通过510(k)的。
-实质等同 Substantial Equivalence
实质等同指的是该申请的产品与已上市产品具有同样的安全性和有效性,即:
- 具有相同的预期用途;且具有相同的技术参数;
或
具有相同的预期用途; 且与之对比后技术参数的不同没有产生新的安全性和有效性的问题;且递交给FDA的资料可以证明此产品至少和已上市产品具有相同的安全性和有效性。
实质等同的声明不需要证明申请产品和对照产品是完全等同的,而可以通过对比预期用途,设计,作用机理,材料,生产过程,性能等参数来建立。
-PMA (上市前批准)
大多数III类设备需要PMA。PMA是严格的上市前提交类型。在FDA批准PMA之前,申办者必须提供有效的科学证据,以证明对设备预期用途的安全性和有效性的合理保证。
PMA通常需要提供临床试验的资料,并且在审批前需要经过FDA的现场审核。
-De Novo (上市前批准)
De Novo提供了一种在没有有效的等同产品的情况下将符合条件的新设备分类为I类或II类的方法。
-HDE (人道主义设备豁免)
HDE为旨在使患有罕见疾病或病症的患者受益的III类设备提供了一条监管途径。为了使设备符合HDE的资格,申办者必须获得人道主义使用设备(HUD)的指定,该名称是通过向FDA的孤儿产品开发办公室(OOPD)申请而获得的。
-上市前提交的信息
根据选择的不同的上市前申请的路径,准备相应的资料包括:
产品描述:
型号规格
工作原理
结构组成
技术指标
预期用途
临床前评价
临床资料
标签,说明书临床资料
……
注:提供测试报告的实验室需要符合GLP的认证
-FDA年费及审核费

-eCopy 电子副本
上市前提交的内容必须包含电子副本(eCopy),可以以光盘(CD),数字视频光盘(DVD)或闪存驱动器作为电子副本的存储媒介。

-510(k)审核时间流程图

上市后的监管
-21 CFR Part 820 C Quality System Regulation
制造商必须建立并遵循质量体系,以帮助确保其产品始终符合适用的要求和规格。FDA管制产品(食品,药品,生物制品和设备)的质量体系被称为当前的良好生产规范(CGMP)。联邦食品,药品和化妆品法案首先授权了820部分(21 CFR 820部分)中设备的CGMP要求。该规定于1978年12月18日生效,并根据第820部分进行了编纂。
QS法规适用于打算商业销售医疗器械的成品器械制造商。
FDA已确定某些类型的医疗设备不受GMP要求。这些设备不受《联邦公报》中公布并在21 CFR 862至892中编纂的FDA分类法规的豁免。免除GMP要求并不免除成品设备制造商保留投诉文件(21 CFR 820.198)或有关记录保存的一般要求( 21 CFR 820.180)。

-医疗器械事故报告
医疗设备报告(MDR)法规(21 CFR第803部分)包含对制造商,设备用户的强制性要求,以便向FDA报告某些与设备有关的不良事件和产品问题。该法规规定,报告应以FDA的Medwatch表格3500A或电子等效文件形式归档。FDA在2014年2月14日发布了规则,要求制造商以电子格式向FDA提交MDR,以便FDA可以对其进行处理,审查和存档。该规定将于2015年8月14日生效。

-汇报时限

- 医疗器械的纠正和移除
根据21 CFR 806,医疗器械的纠正和移除,制造商必须向FDA报告医疗器械的任何纠正或移除,前提是该纠正或移除是为了减少医疗器械引起的健康风险。或纠正由该设备引起的违反本法的行为,而该行为可能会危害健康。

-UDI
UDI规则要求设备标签商(通常是制造商)必须:
在设备标签,设备包装上,有时在设备上直接包含在FDA认可的发行机构的UDI系统下发布的设备标识符(UDI)。
将设备信息提交到设备标识数据库(GUDID)。

总结
选择合适的上市路径,提交合适的文件
考虑多个上市地的要求,提前规划,节约资源
和FDA及时沟通,及时获得反馈
注意尽早的符合体系相关的要求
美德氏医械服务范围
质量管理体系服务
| 服务 | 内容 |
| ISO13485 | ISO 13485的质量体系的培训、建立及运行辅导 |
| FDA | FDA CFR820 的质量体系的培训、建立及运行辅导 |
| MDSAP | MDSAP的质量体系的培训、建立及运行辅导 |
| NMPA | NMPA(GMP、GSP)的质量体系的培训、建立及运行辅导 |
| 质量体系日常维护服务 | FDA820的不符合项以及警告信、欧盟的CAPA、流程改进、质量体系维护的外包、供应商审核 |
| 服务 | 内容 |
| 欧盟市场准入整体解决方案 | 包括CE技术文档撰写、辅导、测试、认证全套方案。还包括欧代服务、欧盟FSC、ISO14971 风险分析、临床评价、灭菌、软件周期、可用性等欧盟合规的咨询与服务 |
| 美国市场准入整体解决方案 | 包括510K文档撰写与认证,产品列名、工厂注册、美国代理人、UDI的合规咨询与服务 |
| 中国市场准入整体解决方案 | 包括NMPA文档撰写与注册,生产许可证、中国FSC、医疗器械广告审核的合规咨询与服务 |
| 其他国家的认证注册咨询服务 | 包括全球法规注册咨询服务,如澳大利亚、新西兰、加拿大、巴西、俄罗斯、日本、韩国等全球国家的注册咨询服务 |
| 服务 | 内容 |
| 国内外法规培训精讲 | MDR 2017/745/EU 法规培训 |
| IVDR 2017/746/EU 法规培训 | |
| MDR临床实验方案设计培训 | |
| IVDR临床实验,性能实验方案设计培训 | |
| ISO14971-2019 医疗器械风险管理培训 | |
| 《ISO13485-2016医疗器械 质量管理体系用于法规的要求》深度解读 | |
| 产品设计开发,产品可用性工程培训 | |
| ISO 11135 11137 灭菌培训 | |
| 其他定制式企业培训 |